ملخص

مقدمة
بيتا ثالاسيميا الصغرى (Beta Thalassemia Minor) ، وتُعرف أيضاً باسم سمة الثالاسيميا (Beta Thalassemia Trait) أو الحامل للمرض (Carrier) .- هي حالة وراثية تنتج عن وجود طفرة في جين واحد فقط من جينات "بيتا غلوبين" المسؤولة عن الهيموجلوبين، بينما يكون الجين الآخر سليماً.
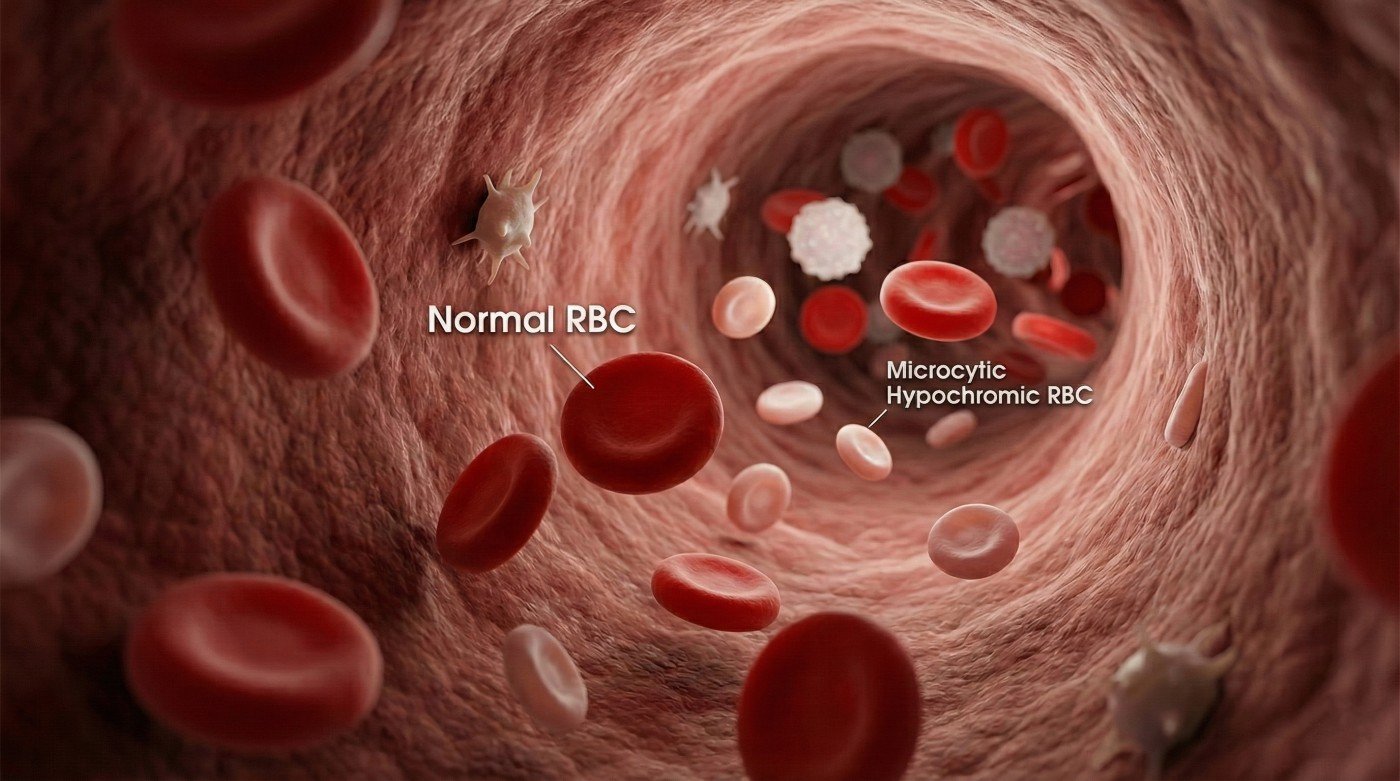
- غالباً ما يكون الأشخاص المصابون بدون أعراض أو يعانون من فقر دم (أنيميا) خفيف جداً لا يتطلب علاجاً أو نقل دم، ويتميز بصغر حجم كريات الدم الحمراء (Microcytosis).
- يتم تأكيد التشخيص مخبرياً عند ملاحظة ارتفاع نسبة الهيموجلوبين A2 (عادة > 3.5%) مع مخزون حديد طبيعي، مما يميزها عن فقر الدم الناتج عن نقص الحديد.
- يجب على المصابين تجنب تناول مكملات الحديد ما لم يثبت وجود نقص فعلي فيه لتجنب تراكمه في الأعضاء، بينما تُنصح الحوامل بتناول حمض الفوليك (5 ملغ يومياً) لدعم إنتاج الدم.
- تكمن الأهمية الرئيسية لهذه الحالة في الوراثة، حيث أن زواج شخصين حاملين للسمة يحمل خطراً بنسبة 25% في كل حمل لإنجاب طفل مصاب ببيتا ثالاسيميا الكبرى (Major) التي تتطلب علاجاً مدى الحياة.
العلامات والأعراض (Signs and Symptoms)
-
الأعراض السريرية (Clinical Symptoms)
في الغالبية العظمى من الحالات، يكون الأشخاص المصابون ببيتا ثلاسيميا الصغرى بدون أعراض (Asymptomatic) ولا يدركون إصابتهم إلا من خلال الفحوصات المخبرية. ومع ذلك، قد تظهر بعض الأعراض الخفيفة المرتبطة بفقر الدم، وتشمل:
-
التعب والإرهاق:
الشعور بالتعب أو الضعف العام هو أكثر الأعراض شيوعًا إذا ظهرت أعراض.
-
شحوب الجلد:
قد يلاحظ شحوب في لون البشرة.
-
ضيق التنفس:
-
الدوار والدوخة:
-
أعراض أخرى محتملة:
قد يعاني المصاب من قصر النفس، خاصةً مع المجهود.
الشعور بالدوار قد يكون عرضًا محتملًا.
في حالات أقل شيوعًا، قد تشمل الأعراض تشنجات (cramps)، خفقان القلب، والصداع المتكرر. كما ذكرت بعض المصادر احتمالية حدوث ضعف في الشهية أو تأخر بسيط في النمو أو انتفاخ البطن، لكنها أكدت أن هذه الأعراض تكون أقل حدة بكثير مقارنة بالثالاسيميا الكبرى.
-
-
العلامات المخبرية والدموية (Hematological Signs)
تعتبر التغيرات في فحص الدم هي العلامة الأساسية لتشخيص الحالة، حيث تظهر النتائج عادةً ما يلي:
-
فقر دم خفيف (Mild Anemia):
يكون مستوى الهيموجلوبين منخفضًا قليلاً أو طبيعيًا، وعادة ما يكون أعلى من 10 غرام/ديسيلتر.
-
صغر حجم كريات الدم (Microcytosis):
انخفاض في متوسط حجم الكرية (MCV) بحيث تكون أقل من 80 فيمتولتر (fL).
-
نقص الصباغ (Hypochromia):
انخفاض في محتوى الهيموجلوبين في الكرية (MCH) بحيث تكون أقل من 27 بيكوغرام (pg).
-
ارتفاع عدد كريات الدم الحمراء (Erythrocytosis):
على عكس فقر الدم الناتج عن نقص الحديد، غالبًا ما يكون عدد كريات الدم الحمراء (RBC count) طبيعيًا أو مرتفعًا (أكثر من 5 مليون/ميكرولتر) في حاملي السمة.
-
تغيرات في شكل الخلايا:
قد يُظهر مسح الدم المحيطي (Peripheral Smear) وجود "خلايا هدفية" (Target cells)، وخلايا بيضاوية (elliptocytosis)، وتفاوت في أحجام وأشكال الخلايا (anisopoikilocytosis).
-
تحليل الهيموجلوبين:
العلامة التشخيصية الفارقة هي ارتفاع نسبة هيموجلوبين A2 (HbA2) عن المعدل الطبيعي (عادة > 3.5%). كما قد يكون هناك ارتفاع طفيف في الهيموجلوبين الجنيني (HbF).
-
-
مضاعفات وحالات خاصة
-
أثناء الحمل:
قد تتفاقم أعراض فقر الدم وتصبح أكثر وضوحًا لدى النساء الحوامل المصابات بالسمة، مما قد يستدعي مراقبة وعلاجًا داعمًا. -
زيادة الحديد:
على الرغم من ندرة حدوث ذلك مقارنة بالأنواع الأشد، إلا أن بعض الدراسات أشارت إلى إمكانية حدوث زيادة في الحديد (Iron Overload) لدى حاملي السمة، خاصة مع تقدم العمر أو وجود عوامل خطر أخرى، نتيجة لزيادة امتصاص الحديد أو خلل في تنظيم الهيبسيدين. -
التشخيص الخاطئ:
غالبًا ما يتم الخلط بين بيتا ثلاسيميا الصغرى وبين فقر الدم الناتج عن نقص الحديد بسبب تشابه صغر حجم الكريات، مما قد يؤدي إلى وصف مكملات الحديد للمريض دون حاجة، وهو أمر قد يكون ضاراً.
بشكل عام، يتمتع الأشخاص المصابون ببيتا ثالاسيميا الصغرى بمتوسط عمر طبيعي ولا يحتاجون عادةً إلى علاج طبي محدد أو نقل دم.
-
الأسباب وعوامل الخطورة
تُعزى أسباب وعوامل خطورة بيتا ثلاسيميا الصغرى، بشكل أساسي إلى عوامل وراثية وجينية محددة، وترتبط ارتباطًا وثيقًا بالأصول العرقية والتاريخ العائلي. فيما يلي تفصيل للأسباب وعوامل الخطورة بناءً على المصادر:
-
الأسباب الجينية (المسببات)
بيتا ثالاسيميا الصغرى هي اضطراب وراثي ناتج عن خلل في الجينات المسؤولة عن إنتاج الهيموجلوبين، وتحدث تحديدًا بسبب:
-
الطفرات الجينية:
تحدث الحالة بسبب طفرات (تغيرات) في جين HBB الموجود على الكروموسوم 11. هناك أكثر من 200 طفرة مختلفة تم تحديدها يمكن أن تسبب هذا الخلل، وتؤثر هذه الطفرات عادةً على عملية نسخ الجين أو ترجمته أو معالجته.
-
الوراثة غير المتماثلة (Heterozygosity):
بيتا ثالاسيميا الصغرى تنتج عن وراثة جين "بيتا غلوبين" معيب من أحد الوالدين فقط، بينما يكون الجين الموروث من الوالد الآخر طبيعيًا. هذا يختلف عن "بيتا ثالاسيميا الكبرى" التي تتطلب وراثة جينين معيبين (واحد من كل والد).
-
نقص تخليق السلاسل:
تؤدي الطفرة إلى نقص (يُرمز له بـ +β) أو غياب كامل (يُرمز له بـ β0 ) في إنتاج سلاسل "بيتا غلوبين" من الجين المصاب. وجود جين واحد سليم يسمح بإنتاج كمية كافية من الهيموجلوبين ليعيش الشخص حياة طبيعية دون فقر دم شديد، ولكنه يؤدي إلى صغر حجم كريات الدم الحمراء (Microcytosis).
-
-
عوامل الخطورة
تزيد العوامل التالية من احتمالية أن يكون الشخص حاملًا لسمة بيتا ثالاسيميا:
-
التاريخ العائلي والوراثة:
يعتبر وجود تاريخ عائلي للإصابة بالثالاسيميا هو عامل الخطر الرئيسي، حيث ينتقل المرض من الآباء إلى الأبناء عن طريق الجينات. إذا كان أحد الوالدين حاملًا للسمة (بيتا ثالاسيميا الصغرى) والآخر سليمًا، فإن هناك احتمالية بنسبة 50% في كل حمل أن يكون الطفل حاملًا للسمة أيضًا.
-
ب. العرق والأصول الجغرافية:
ترتفع معدلات انتشار جينات بيتا ثالاسيميا بشكل ملحوظ في مجموعات عرقية ومناطق جغرافية معينة،ويرتبط ذلك تاريخيًا بالمناطق التي تنتشر فيها الملاريا. تشمل المجموعات الأكثر عرضة للخطر:
-
سكان حوض البحر الأبيض المتوسط:
مثل الإيطاليين (خاصة في صقلية وسردينيا)، واليونانيين، وسكان قبرص. في قبرص، تصل نسبة حامليالسمة إلى 15%.
-
الشرق الأوسط:
بما في ذلك إيران، العراق، وسوريا. في إيران، تصل النسبة إلى 10% في المناطق الجنوبية والشمالية.
-
جنوب شرق آسيا:
مثل تايلاند والصين (خاصة الجنوب).
-
إفريقيا:
الأفراد من أصول أفريقية.
-
شبه القارة الهندية.
-
-
الملاريا:
يُعتقد أن الانتشار العالي لهذه الطفرات الجينية في المناطق المذكورة أعلاه ناتج عن "ميزة البقاء" (Selective Advantage)؛ حيث أن حاملي سمة الثالاسيميا يكونون أكثر مقاومة للإصابة بالملاريا المنجلية (Plasmodium falciparum) مقارنة بالأشخاص الأصحاء تمامًا، مما سمح للجين بالبقاء والانتشار عبر الأجيال في تلك المناطق الموبوءة.
-
-
حالات جينية معقدة وتحديات التشخيص
في بعض الحالات، قد تتأثر دقة تشخيص "بيتا ثالاسيميا الصغرى" بوجود عوامل جينية أخرى، مما يشكلتحديًا في تحديد الحالة:
-
الثالاسيميا المقنعة (Masked Thalassemia):
قد يحمل الشخص طفرة بيتا ثالاسيميا ولكنه يظهر مستويات طبيعية من الهيموجلوبين A2 (وهو العلامة التشخيصية المعتادة)، وذلك بسبب وجود طفرة متزامنة في جين "دلتا غلوبين" (HBD)، مما يخفي التشخيص التقليدي.
-
نقص الحديد:
يمكن لنقص الحديد الشديد أن يخفض مستويات HbA2، مما قد يؤدي إلى عدم تشخيص بيتا ثالاسيميا الصغرى لدى الحاملين لها.
-
الطفرات الصامتة:
بعض الطفرات (Silent variants) تؤدي إلى تأثيرات طفيفة جدًا لدرجة أن التحاليل الروتينية قد لا تكتشفها، ويكون الشخص "حاملاً صامتًا".
-
العلاج (Treatment)
في الغالبية العظمى من الحالات، لا يحتاج الأشخاص المصابون بـ بيتا ثالاثيميا الصغرى أو "السمة" إلى أي علاج طبي محدد، حيث تكون الحالة عادةً بدون أعراض أو تسبب فقر دم خفيفاً جداً لا يؤثر على الحياة اليومية. ومع ذلك، هناك اعتبارات علاجية ووقائية مهمة يجب اتباعها لإدارة الحالة بشكل صحيح وتجنب المضاعفات، خاصة أثناء الحمل أو في حالات زيادة الحديد.
إليك التفاصيل العلاجية والإدارية بناءً على المصادر:-
الإدارة العامة وتجنب العلاجات الخاطئة
-
لا حاجة للعلاج الروتيني:
الأشخاص المصابون بسمة الثالاثيميا لديهم متوسط عمر طبيعي ولا يحتاجون عادةً إلى مراقبة طبية طويلة الأمد أو تدخلات علاجية. -
تجنب مكملات الحديد:
الخطأ الطبي الأكثر شيوعاً هو تشخيص الحالة خطأً على أنها فقر دم بسبب نقص الحديد ووصف مكملات الحديد. يجب تجنب تناول الحديد ما لم يثبت وجود نقص فعلي في مخزون الحديد (الفيريتين) عبر التحاليل المخبرية، لأن تناول الحديد دون حاجة قد يؤدي إلى تراكمه في الجسم وتضرر الأعضاء. -
تجنب التشخيص الخاطئ:
نظراً لأن الحالة تسبب فقر دم ذو كريات صغيرة (Microcytic Anemia)، يجب تمييزها عن نقص الحديد لتجنب العلاجات غير الضرورية. التشخيص الدقيق يتطلب تحاليل مثل فصل الهيموجلوبين (Hb electrophoresis) وقياس الفيريتين.
-
-
المكملات الغذائية (حمض الفوليك)
-
في الحالات العامة:
لا تشير الدلائل الحالية بوضوح إلى ضرورة تناول حمض الفوليك بشكل روتيني لجميع المصابين بالسمة الذين ليسوا حوامل ولا يخططون للحمل، حيث أن تمدد نخاع العظم لديهم أقل بكثير مقارنة بالثالاثيميا الكبرى. ومع ذلك، قد يُوصف في حالات فقر الدم الخفيف لدعم إنتاج خلايا الدم الحمراء. -
أثناء الحمل والتخطيط له:
يُوصى بشدة للنساء المصابات ببيتا ثالاثيميا الصغرى بتناول مكملات حمض الفوليك بجرعة 5 ملغ يومياً (وهي جرعة أعلى من المعتاد للنساء غير المصابات)، وذلك بدءاً من ثلاثة أشهر قبل الحمل وطوال فترة الحمل، لمنع تفاقم فقر الدم ودعم نمو الجنين.
-
-
إدارة زيادة الحديد (Iron Overload)
-
الفصد المنخفض الشدة (Mini-phlebotomies):
في حالات زيادة الحديد المؤكدة لدى حاملي السمة، قد لا يكون الفصد التقليدي (سحب كميات كبيرة من الدم) مناسباً بسبب فقر الدم الخفيف لديهم. لذا، اقترحت دراسات حديثة استخدام "الفصد المصغر" (سحب 150-350 مل من الدم) كل أسبوعين إلى ثلاثة أسابيع كخيار علاجي آمن وفعال لتقليل الفيريتين دون تفاقم فقر الدم. -
مخلبات الحديد (Iron Chelators):
لا توجد إرشادات رسمية لاستخدام أدوية طرد الحديد (مثل ديفيراسيروكس - deferasirox أو ديفيريبرون - deferiprone) لحاملي السمة، واستخدامها قد يكون غير مصرح به (off-label) أو يحمل مخاطر لبعض المرضى، لذا يتم اللجوء إليها بحذر شديد وفي حالات خاصة جداً. -
تعديل نمط الحياة:
يُنصح المرضى الذين يعانون من زيادة الحديد بتجنب الكحول والسيطرة على العوامل الأيضية (مثل السمنة وارتفاع السكر) لأنها تفاقم تراكم الحديد وتضرر الكبد.
على الرغم من ندرتها مقارنة بالثالاثيميا الكبرى، قد يعاني بعض حاملي السمة من زيادة في الحديد (Hyperferritinemia) نتيجة عوامل متعددة مثل زيادة الامتصاص المعوي للحديد بسبب خلل تنظيم الهيبسيدين، أو عوامل أيضية أخرى،. تشمل الخيارات العلاجية في هذه الحالات الخاصة ما يلي:
-
-
الإدارة أثناء الحمل
-
المراقبة:
قد ينخفض الهيموجلوبين بشكل ملحوظ أثناء الحمل، مما قد يستدعي نقل الدم في حالات نادرة إذا كان فقر الدم شديداً ويؤثر على نمو الجنين، رغم أن هذا غير شائع لحاملي السمة. -
الفحوصات:
يُنصح بإجراء فحوصات للقلب والكبد والغدة الدرقية قبل الحمل للتأكد من عدم وجود مضاعفات ناتجة عن تراكم الحديد، خاصة إذا كان هناك تاريخ لنقل الدم. -
إيقاف بعض الأدوية:
إذا كانت المريضة تتناول مخلبات الحديد (في حالات نادرة)، يجب إيقافها قبل الحمل بثلاثة أشهر لأنها قد تؤثر على الجنين.
تحتاج المرأة الحامل المصابة ببيتا ثالاثيميا الصغرى إلى رعاية خاصة:
-
الوقاية (Prevention)
بما أن الحالة وراثية، فإن الخيارات الوقائية تتركز حول التخطيط الإنجابي والاستشارة الوراثية:
-
الاستشارة الوراثية والفحص قبل الزواج:
ينصح بشدة بإجراء فحوصات قبل الزواج أو الحمل لمعرفة ما إذا كان الشريك أيضاً حاملاً للسمة. إذا كان كلا الوالدين حاملين للسمة، هناك خطر بنسبة 25% لإنجاب طفل مصاب بـ "بيتا ثالاسيميا الكبرى" (النوع الخطير)، و50% أن يكون الطفل حاملاً للسمة (صغرى).
-
التشخيص الوراثي قبل الزرع (PGD):
بالنسبة للأزواج المعرضين لخطر نقل الثالاسيميا، يمكن استخدام تقنيات الإخصاب في المختبر (IVF). يتم في هذه العملية فحص الأجنة جينياً قبل زرعها في الرحم لاختيار الأجنة التي لا تحمل الطفرة الجينية المسببة للثالاسيميا، وبذلك يمكن "الوقاية" من ولادة طفل مصاب أو حامل للسمة.
مصادر تم الاستعانة بها
- دليل الاستشارات الطبية حول الثلاسيميا – ARUP Consult.
- كتاب مرجعي شامل: StatPearls - الثلاسيميا – المكتبة الوطنية للطب (NCBI).
- الجرعة الموصى بها ومدة العلاج بحمض الفوليك – Dr. Oracle.
- معدل العمر المتوقع لمريض الثلاسيميا – مستشفى Liv.
- أعراض وعلامات مرض الثلاسيميا – City of Hope.
- دراسة بحثية حول إدارة مرض الثلاسيميا – مركز PubMed Central.
- بحث حول تراكم الحديد وعلاجه في الثلاسيميا – مركز PubMed Central.
- المضاعفات طويلة الأمد لمرضى الثلاسيميا بيتا – مركز PubMed Central.
- استراتيجيات علاجية حديثة للثلاسيميا – مركز PubMed Central.
- ما هي الثلاسيميا بيتا؟ (معلومات شاملة) – جامعة كاليفونيا UCSF.
- مراجعة سريرية: الثلاسيميا بيتا – المكتبة الوطنية للطب (NCBI).
- مراجعة سريرية: الثلاسيميا ألفا – المكتبة الوطنية للطب (NCBI).
- نظرة عامة على أمراض الثلاسيميا – كليفلاند كلينك.
- حالة نادرة: الثلاسيميا المقنعة (دراسة) – ResearchGate.
- سمة الثلاسيميا بيتا (الثلاسيميا الصغرى) عند الأطفال – مستشفى Nicklaus Children's.
- تشخيص وعلاج الثلاسيميا – مايو كلينك.
- معلومات صحية حول الثلاسيميا بيتا – ستانفورد للأطفال.
- ورقة بحثية حول الثلاسيميا (ملف PDF) – OA Text.
- تفاصيل موسعة حول الثلاسيميا بيتا – كليفلاند كلينك.
- مقال علمي متخصص في أمراض الدم – مجلة Haematologica.
- إرشادات الثلاسيميا بيتا والحمل (ملف PDF) – هيئة الخدمات الصحية في ويلز (NHS).
- الفحص الجيني والمخاطر الوراثية للثلاسيميا – مستشفى سانت جود.
- دليل المريض: الثلاسيميا بيتا والحمل – الكلية الملكية لأطباء النساء والتوليد.
- دليل الآباء: التعامل مع الثلاسيميا بيتا – موقع KidsHealth.
- مقالة طبية: التعامل مع اعتلالات الهيموجلوبين – الأكاديمية الأمريكية لأطباء الأسرة.
- مراجعات جينية: الجينات المرتبطة بالثلاسيميا بيتا – GeneReviews (NCBI).
- كيف أعالج الثلاسيميا (بروتوكولات علاجية) – جمعية أمراض الدم الأمريكية (Blood Journal).
- أعراض وأسباب الإصابة بالثلاسيميا – مايو كلينك.